Les récentes émergences de SARS-CoV (2003), MERS-CoV (2013) et SARS-CoV-2 (2020) ont démontré le haut potentiel épidémique des coronavirus. De nombreuses souches isolées chez les chauves-souris ou les pangolins sont très proches du SARS-CoV-2 (>96% d’homologie pour RaTG13), ce qui indique une origine animale de la pandémie COVID-19. Pourtant, les mécanismes responsables du franchissement de la barrière d’espèces de l’animal à l’homme sont inconnus.
Des chercheurs américains (Regional College of Veterinary Medicine, Blacksburg) ont recherché dans le génome du SARS-CoV-2 les signatures moléculaires d’une sélection positive qui pourraient être à l’origine de ce franchissement. En utilisant de puissants algorithmes statistiques (OmegaPlus et RAiSD), ils ont analysé 182 792 génomes viraux référencés dans la base GISAID. Ils ont alors identifié les régions portant des mutations qui se sont propagées mais qui ont peu évoluées (c’est ce qu’on appelle un « balayage sélectif »).
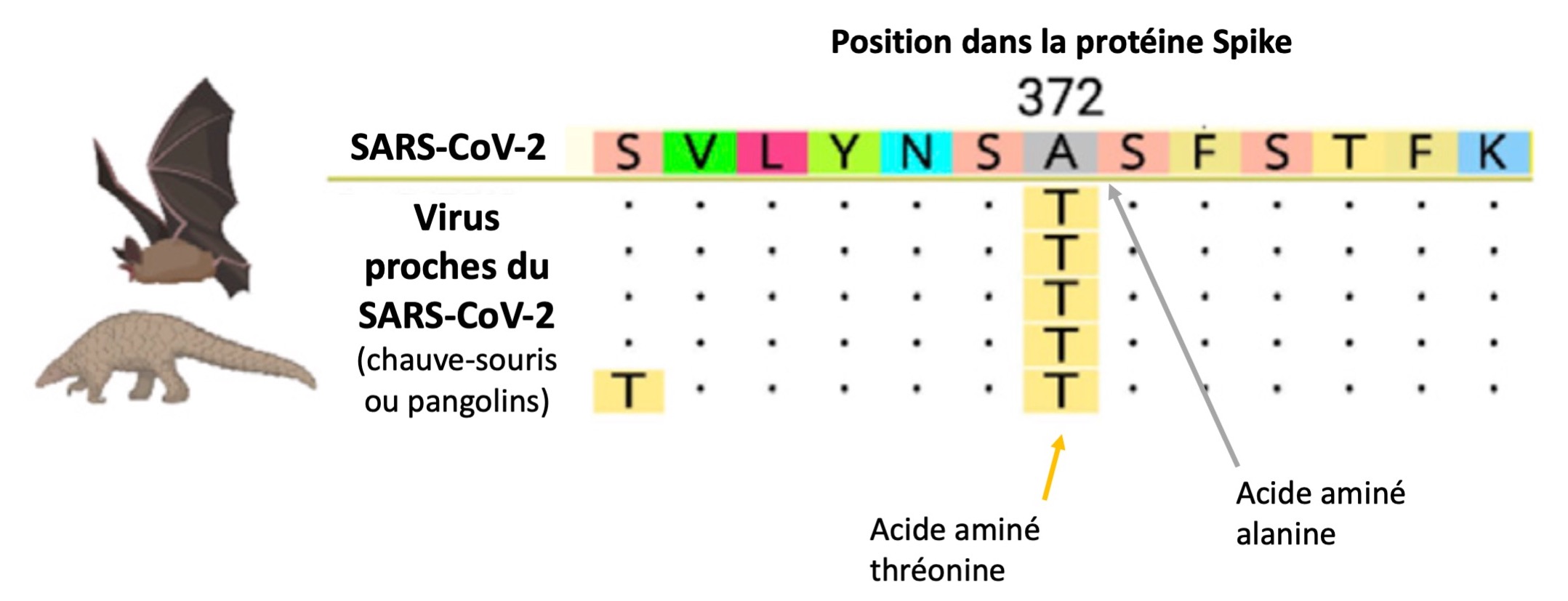
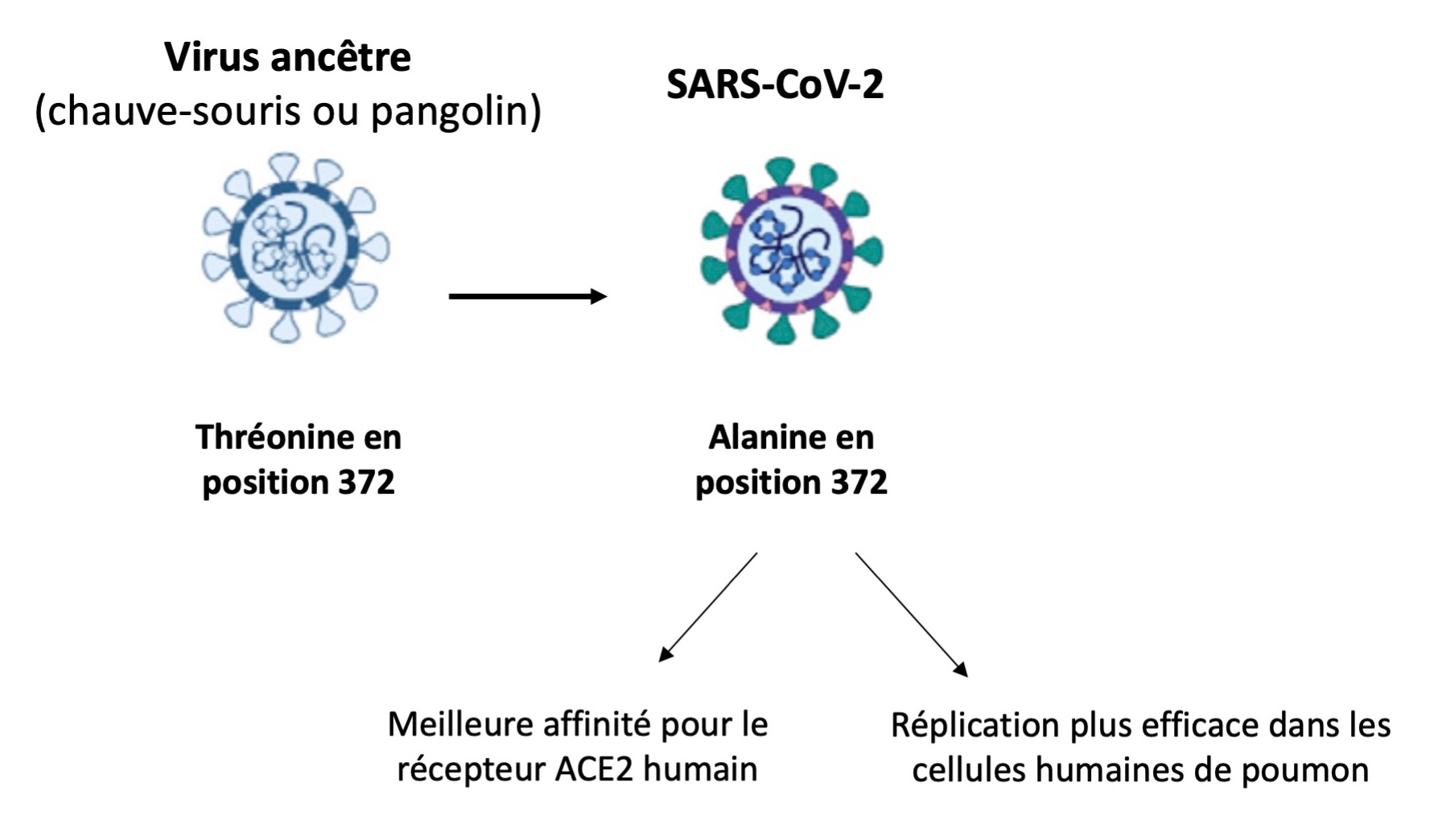
Parmi 8 régions génomiques détectées, 4 sont localisées dans le gène ORF1ab et 4 dans celui de Spike, la protéine qui régit l’entrée dans les cellules humaines et donc l’ensemble des cibles du virus (ou son « tropisme »). Il faut savoir que le SARS-CoV-2 fait partie des sarbecovirus (sous-genre des β-coronavirus). Il est donc intéressant de chercher les différences entre eux pour retracer son histoire génétique.
Une différence importante est alors apparue. Ils ont identifié un acide aminé de la protéine Spike comme étant différent d’un virus à d’autres. Ce « résidu » 372, situé dans le RBD de Spike (Receptor Binding Domain), est en effet une thréonine (T) chez les sarbecovirus, mais une alanine (A) dans les SARS-CoV-2 humains. Ce sont donc des acides aminés différents issus d’une mutation.