SARS-CoV-2 belongs to the family of Beta-coronaviruses. Out of the 7 viruses from this family that infect humans, two have already been responsible for epidemics and are relatively close to SARS-CoV-2 (SARS-CoV-1 and MERS-CoV), while the others are seasonal viruses causing respiratory infections. SARS-CoV-2 is an enveloped virus, with a single-stranded RNA genome (one of the largest genomes among RNA viruses).
On the surface of the virus we find the spike (S) protein, the key component and principal target of vaccines currently in development. This surface viral protein involved in binding and entry into the cell has flexible and mobile domains that enable significant changes in conformation. Between the mobile parts there are points that remain fixed in their conformation. Before fusion with the cellular membrane, the S proteins form trimers in the shape of crowns. Each S protein is made up of 2 domains (S1 and S2), delimited by a cleavage site recognized by the cellular protease furine (SD2): in other words the furine can divide the S into 2, thereby forming the domains S1 and S2. The S1 domain includes a particular region that binds to the ACE2 cellular receptor and is called the RBD (Binding Domain Receptor). The S2 domain includes a second cleavage site (S2’), as well as a peptide fusion, enabling the fusion of viral and cellular membranes when the viral particle enters the cell. This S2 domain undergoes important conformation changes at the time of the cleavage of its S2’ site by the cellular protease TMPRSS2, present on the surface of the endothelial cells.
Although the rate of mutation of the SARS-CoV-2 genome is very low compared to other RNA viruses, the regular emergence of variants presenting mutations has been observed, notably in the spike protein. This is the case with the D614G mutation (aspartic acid becomes a glycine in position 614) which has become, since summer 2020, a majority variant throughout the world, giving us the so-called “English” and “South African” variants recently identified.
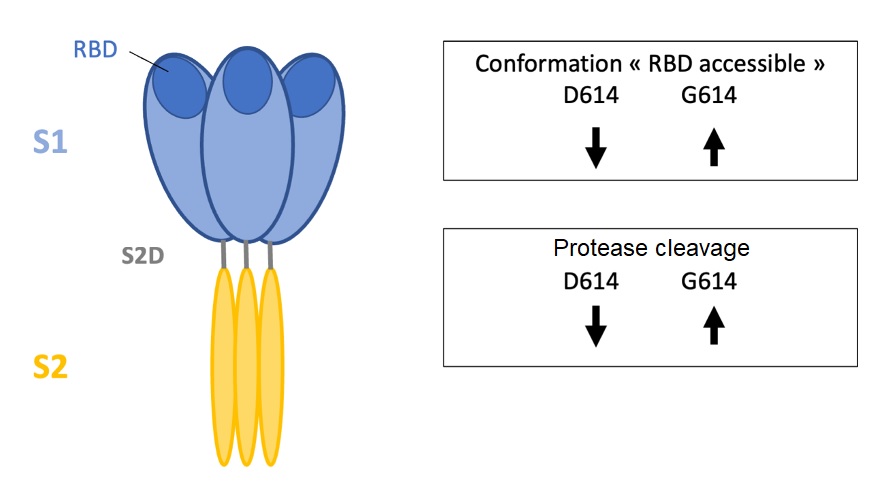
In order to study the conformation of the S protein, researchers at Durham in the USA obtained, through genetic engineering, soluble forms of the mutants studied (D614 et G614). This modification of the protein enables it to be kept in pre-fusion conformation and to block the conformational transition to the post-fusion form. The impact of the D614G mutation on the structure and the antigenicity of the S1 domain was first analyzed using electron cryomicroscopy. This technique allows the study of proteins, and confirmed that the D614G mutation is situated in a key structural region. Two S protein populations were identified: one conformation that makes the RBD domain accessible to enable binding to the ACE2 cellular receptor, and one conformation making the RBD domain inaccessible. It was observed that the D614G mutation modifies the ratio of these two conformations. This shows that a single mutation in this region may lead to important modifications in the protein’s structure and its conformation.
The mutation of the residue aspartic acid into glycine therefore modifies the equilibrium observed between the S protein conformations (RBD accessible or non-accessible). The proximity of this mutation to the SD2 cleavage domain separating the S1 and S2 domains prompted scientists to examine the effect of this mutation on the protein cleavage. The results could indicate that the D614G mutation increases the probability of cell protease cleaving the S1/S2 junction. This could then entail an increase in the replication and transmissibility of the virus. The scientists also showed that changes in the S2 domain can strongly impact on the conformation of the RBD domain.