The appearance and rapid spread of SARS-CoV-2 variants may have adverse effects on the effectiveness of current vaccines. It is therefore crucial to identify new variants quickly, and to sequence them, that is, to read the succession of letters that make up their genome.
Today 610 000
SARS-CoV-2 genomes have been sequenced across the world. Amongst these analyses, phylogenetic studies, where the results are generally represented in the form of trees, have proved to be the most important tools to keep track of variants: the closer genomes are, the closer they are in the phylogenetic tree as well. But how do can these analyses enable us to monitor the appearance of new variants before they become dominant across the world and before problems of resistance to vaccines or medicines arise?
- The first useful purpose of these analyses is to help us follow transmission episodes and be able to retrace the origin of groups involved in transmission.
- The second is to be able to monitor key mutations in the viral genome. The appearance or the spreading of a variant carrying a specific mutation may, for example, modify vaccine strategy, if the mutation confers greater vaccine resistance on the virus. Another example is the discovery of combined mutations on mink farms in Denmark that seemed to give the virus greater resistance to antibodies.
- Finally, these analyses enable us to better estimate data linked to the pandemic, notably the R0, a key figure in evaluating the virus’ transmission.
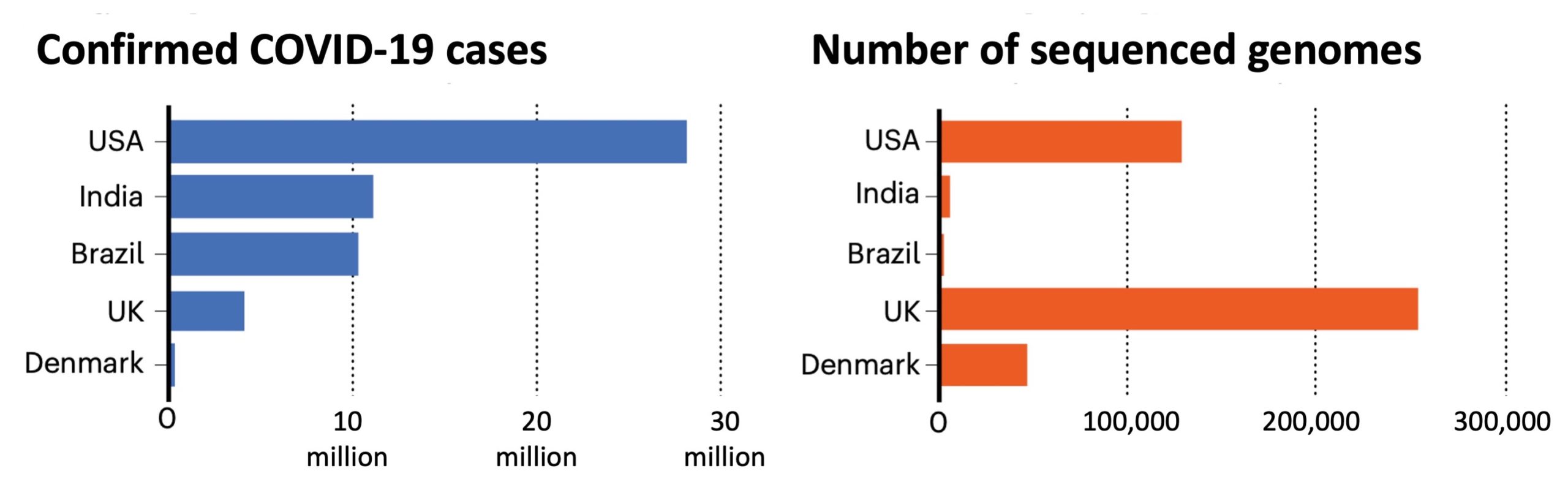
However, one of the hazards of the multiplication of viral genome sequencing is the risk of errors in sequences that are shared online (contaminations, sample errors, poor sequencing quality). The rush to obtain fully analysed sequences by non-expert laboratories greatly increases this risk. In addition, there is great disparity between nations concerning the number of genomes sequenced. The UK has for example sequenced 5000 for every 100 000 cases, the US 320 and Brazil 30. These discrepancies must be taken into account when studying the transmission of variants across the world.